How do we transform raw, extracted DNA into a perfectly prepared sequencing library, ready for advanced analysis?
Just as a chef prepares ingredients before cooking, scientists must prepare DNA before sequencing. Raw DNA, fresh from extraction, isn’t immediately compatible with sequencing instruments. This lesson walks through the essential, meticulous steps of Oxford Nanopore library preparation: end repair, dA-tailing, barcoding, and pooling. These processes are not just technical; they are fundamental transformations that make high-throughput DNA sequencing possible, allowing us to unlock the genetic secrets of entire microbial communities.
The Crucial First Step: Why Library Prep?
Sequencing instruments cannot simply read raw extracted DNA. The DNA must first be prepared into a sequencing library.
Why do you think raw DNA isn’t directly compatible with sequencing machines? What characteristics might make it unsuitable?
Precision and Planning: Input Calculation
The protocol begins by calculating how much sample volume contains 400 nanograms of DNA. Divide the DNA needed by the DNA concentration. If the sample is 200 nanograms per microliter, 400 divided by 200 equals 2 microliters.
The amount of DNA present in a given volume, typically measured in nanograms per microliter (ng/µL). Precise measurement of DNA concentration is fundamental for molecular biology experiments to ensure correct input amounts.
Refining the Ends: End Repair and dA-Tailing
End repair fixes damaged or incompatible DNA ends. dA-tailing adds a single adenine overhang, preparing the DNA to accept barcode adapters.
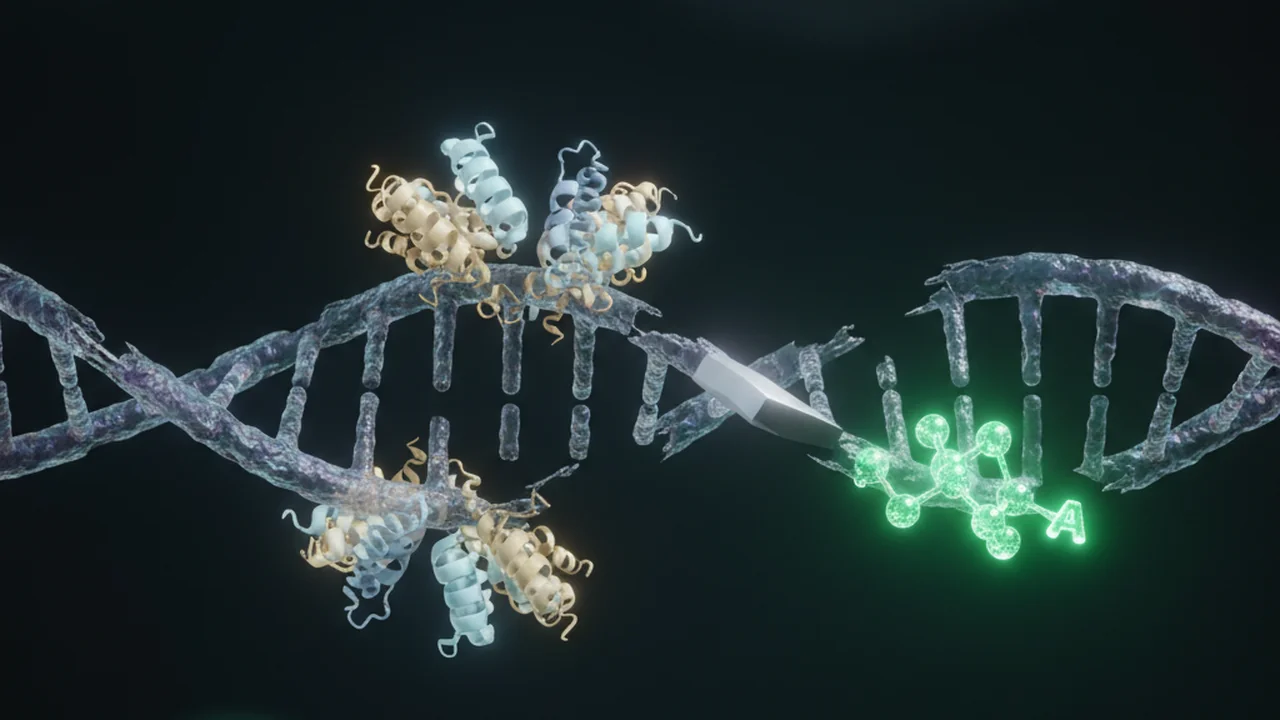
The 5′-phosphate group added during end repair is essential for the DNA ligase enzyme to form a phosphodiester bond, covalently linking the DNA fragments to the adapters in subsequent steps.
All extracted DNA fragments have identical, blunt ends ready for immediate attachment.
DNA extraction and fragmentation can create a variety of ends (blunt, sticky, nicks). End repair and dA-tailing are enzymatic processes that standardize these ends, creating a uniform substrate for adapter ligation.
The Workflow: Reaction Setup and Cleanup
The reaction combines DNA, end repair/dA-tail master mix, and water to a total volume of twelve microliters. After incubation, magnetic bead cleanup removes unwanted reaction components while retaining DNA.

You need to set up 10 individual library preparation reactions. Each reaction requires 400 ng of DNA, 5 µL of master mix, and water to bring the total volume to 12 µL. If your DNA stock is 250 ng/µL, calculate the total volume of DNA, master mix, and water needed for all 10 reactions, plus an extra 10% for pipetting error.
- Calculate the volume of DNA needed per reaction.
- Calculate the volume of water needed per reaction.
- Multiply each component’s volume by 10 (for 10 reactions).
- Add 10% extra to each total volume to account for pipetting losses.
- Raw DNA must be processed into a sequencing library.
- Accurate input DNA quantification is crucial for successful reactions.
- End repair and dA-tailing prepare DNA ends for adapter ligation.
- Magnetic bead cleanup purifies DNA after enzymatic reactions.
Identity Tags: Barcoding
Each sample receives a unique DNA barcode. This short sequence acts like a molecular ID tag so many students’ samples can be mixed together and sorted later during analysis.

Barcoding is a cornerstone of modern high-throughput sequencing. It allows labs to dramatically scale up their experiments, processing hundreds or thousands of samples in parallel, which is vital for projects like large-scale microbiome studies or population genetics.
Efficiency Amplified: Multiplexing and Pooling
Pooling barcoded samples is called multiplexing. It lets many libraries be sequenced in the same run without losing track of which read came from which sample.

The technique of combining multiple individually barcoded DNA libraries into a single tube for simultaneous sequencing, significantly increasing efficiency and reducing the cost per sample.
Want to go deeper? The design of barcode adapters
Nanopore barcode adapters are specifically engineered DNA constructs. They contain not only the unique barcode sequence but also sequences essential for ligation to the prepared DNA fragments and for binding to the nanopore sequencing platform. The 3′ end of these adapters typically features a T-overhang, which specifically ligates to the A-overhang created during the dA-tailing step. This directed ligation ensures that the adapters attach correctly and efficiently, preparing the DNA for its journey through the nanopores.
Which of the following best describes the primary benefit of multiplexing in DNA sequencing?
Purification Post-Pooling: Pooled Cleanup
After pooling, magnetic beads capture the barcoded DNA library. Washes remove contaminants, brief drying removes ethanol, and elution recovers the cleaned pooled library.
The pooled cleanup involves several critical steps: adding beads, incubation, magnetic separation, washes, drying, and elution. Choose two of these steps and explain the precise reason why each is necessary for obtaining a high-quality sequencing library. What would be the consequence of skipping or incorrectly performing these two steps?
The Final Leg: Bridge to Adapter Ligation
The prepared barcoded pool is now ready for adapter ligation, the final step that enables nanopore sequencing.

Oxford Nanopore library preparation is a systematic process involving precise input calculation, enzymatic modification of DNA ends, unique barcoding, efficient multiplexing, and thorough purification to create a high-quality, sequence-ready DNA library.
Pooling barcoded samples is called multiplexing. It lets many libraries be sequenced in the same run without losing track of which read came from which sample.
The Shift
- Raw DNA requires precise enzymatic modifications, like end repair and dA-tailing, to ensure compatibility with sequencing adapters and prevent errors.
- Barcoding and multiplexing are strategic approaches that dramatically increase the efficiency and cost-effectiveness of sequencing by allowing many samples to be processed concurrently.
- Rigorous procedural steps, including accurate input calculation, careful reaction setup, and multi-stage magnetic bead cleanup, are indispensable for generating high-quality DNA libraries suitable for nanopore sequencing.